Speech by Mark Harrington
Presented at VIII International Conference on AIDS, Amsterdam, July 22, 1992
I would like to dedicate my remarks today to my colleagues in the struggle against AIDS who did not survive the year since this group last met in Florence – to Charles Barber, Daniel Sotomayor, David Byar, David Lopez, Donald Ruddy, Jay Lipner, Jerry Jontz, Jose Perez, Maritza Ramos, Michael Wright, Sonia Singleton, Terry Beirne, Tom Hannan, and particularly to Scott Slutsky. A writer, musician, AIDS activist and long-term survivor, Scott joined the Treatment + Data Committee of ACT UP/New York in spring 1989, worked with us on our first Treatment Research Agenda for the Montreal conference that year, helped to start the Countdown 18 Months Project targeting the five major opportunistic infections, and joined TAG, the Treatment Action Group, when we split off from ACT UP last fall. Scott continued his work through the last weeks and days of his life as he struggled with CMV and CNS lymphoma. He was brave and kind, unflinching and selfless in his work. No one can replace him.
This morning, I’m going to talk about where we in the AIDS community stand after five years of relentless treatment activism, why we need to change, and how you – the researchers and clinicians working full-time on AIDS and HIV infection – can help us to find out what we need to know and do.
Since 1987, the activist critique of AIDS research has worked its way back: from drug approval at the regulatory level of the US Food + Drug Administration (FDA), to expanded access for drugs still under study (Parallel Track), to the design and conduct of the controlled clinical trials themselves by the National Institutes of Health (NIH), pharmaceutical companies and, community-based clinical trial centers.
While this work has generated some useful reforms in an inefficient system, it often seems that all these accomplishments go for naught. HIV keeps spreading, AIDS keeps striking people down, and researchers appear to have little confidence in the rapid development of a therapeutic cure or an effective vaccine. Against a background of deepening political reaction, declining research subsidies, and pervasive pessimism about the prospects for a scientific breakthrough, some activists have grown unsure of the value of continuing to engage the scientific infrastructure. What is the point of streamlining access and approval when the result is merely to replace AZT with additional mediocre, toxic, expensive nucleoside analogues? What is the point of developing prophylaxis and better treatment for opportunistic infections when these measures simply allow someone to survive long enough to develop lymphoma, visceral Kaposi’s sarcoma, wasting syndrome or neuropathology?
We still lack definitive answers to the simplest and most crucial questions about the foundation of most people’s antiretroviral regimen, AZT. Last month my colleague Mike Barr addressed five such questions to the US National Commission on AIDS:
1. Do we know how the drug might work? No.
2. Do we know when to initiate therapy for maximum clinical benefit? No.
3. Do we know the best dose? No.
4. Do we know if the drug extends life? No.
5. Do we know if a patient is better off taking the drug or not taking it? The answer is, “No. We do not know even this.”
If the reforms won by activists are not to become mere stratagems for unimaginative pharmaceutical companies to swiftly develop and market a whole series of additional nucleoside analogues (d4T, FLT, 3TC, etc.), activists must become more involved in the basic research process itself, forcing academic and industrial researchers to turn their attention to novel approaches to HIVinduced immune suppression, including immune based therapy, cytokine inhibition, and active immunotherapy, with the ultimate goals of elucidating the pathogenesis of AIDS, stopping its progression, and reversing its damage. The task requires that activists become as familiar with basic research on immunopathogenesis as they have become with the once arcane jargon and methodology of randomized clinical trials.
1. Activists need to become involved in basic research.
Why don’t we have a cure for AIDS? We don’t have a cure for AIDS because we still don’t know how HIV triggers the collapse of the immune system. We know lots about the cycle of HIV in the test tube but virtually nothing about its life cycle in our bodies. Why is the initial immune response so strong, and how does it ultimately fail? Why do some people develop AIDS within a year of infection, while others, 12 years on, remain asymptomatic and immunologically intact? What lessons could be learned (and are not being asked) from studying long-term survivors? What is the significance of early immune defects in responses to recall antigens and alloantigens? How are the thymus and the lymphoid tissues destroyed? How are the T cells killed? Does the immune decline producing AIDS and the final T cell drop occur only after the destruction of the peripheral lymphoid tissues? What are the prospects for reconstruction of the thymus and the lymphoid tissue as well as the prospects for retraining new T lymphoblasts? What is the role of extra-lymphoid infection, e.g., in the lungs, the gut, and the brain? What is the role of APC such as LC, DC, FDC and macrophages either as viral reservoirs or as defective APC in the progression to AIDS? Are the T cells killed by the virus, or by the immune system itself in a misguided effort to eliminate the virus? Are antibodies, complement, CTLs or macrophages the main culprits? Does immune hyperactivation lead to immune exhaustion, T4 cell depletion and AIDS? If so, what are the roles of copathogens and host factors in sustaining immune activation? If so, what is the role of immune deactivating interventions? If signaling defects or programmed cell death cycles are involved in T cell depletion, how can the defects be corrected and apoptosis aborted?
Ask anyone you respect: “Do you really believe that direct cell killing is the major mechanism of immune destruction in HIV infection?” Get them to answer specifically. We need to know. If this mechanism is no longer adequate to explain the disease, get them to admit this. You need to come clean so we can start over, if necessary, with a clear slate. Last year, many researchers started talking about starting over. Some people said “we’ve just finished the first five years of a thirty year campaign.” Great. Others said “It’s back to square one.” When you go back to square one, we go back to ground zero – to the killing fields without hope or help where we continue to die wondering anew whether all the research-sanctioned nucleosides we’ve been stuffing into our bodies for the last five years, alone or in combination or cycling like cancer chemotherapy, have been doing anything at all, or, on balance, anything good.
We don’t have time to start over. In the AIDS community, we’ve only got one shot. I’m here to ask you, the researchers, to take some simple steps to speed up the process of elucidating the pathogenesis of AIDS and developing a cure. (Yes, cure.) Part of that involves asking you a tough question: Do you really believe there is a rational basis for saying that a cure for AIDS is possible? If so, what are the prerequisites? What are the obstacles? What are the most crucial questions which must be answered in order to develop a cure and a preventive vaccine?
2. Basic research need to become more clinical.
Two years ago, David Ho delivered something of a shock to many virologists in San Francisco. His studies showed that wild-type isolates of HIV from infected persons were 100-1000 times less susceptible to inhibition by rsCD4 therapy than were laboratory lines such as HIVHTLVIIIB. By the simple expedient of using clinical isolates, Ho demonstrated why a multi-million dollar effort, involving multiple drug companies and some of America’s top AIDS researchers, had failed. Years and fortunes could have been saved if these experiments had been conducted ahead of time. Since then, many of you have learned a lot more about how artificial laboratory strains of HIV can yield misleading results. Yet much vaccine and therapeutic screening work continues to use laboratory strains. They are convenient, predictable and well-known. (They may also assist in providing a false picture of pathogenetic mechanisms, since some of them kill CD4 cells much more readily than field isolates).
I’m concerned that much time and perhaps many lives could be saved if basic researchers took the basic lesson of Dr. Ho’s maxim: to culture is to disturb. Yes, it is harder to use clinical samples. Yes, it is messier, and yes, it is harder to control. Experiments may be less elegant – but they will be more relevant. How much do we know about what the virus does in the T cells of infected people? Virtually nothing. How much do we know about what the virus does in the macrophages and the dendritic cells of infected people? Very little. How much do we know about what the virus does in the bone marrow, thymus, peripheral lymphoid tissues, mucosal and non-mucosal areas of the body? Next to nothing. How much do we know about how the immune system itself damages cells, tissues, organs and bodies of people with AIDS or HIV? Very little. [In spite of this lack, we still have experts opining on the basis of very little evidence that autoimmune processes do not play a role in AIDS.] For ten years, many of the most prominent and powerful AIDS researchers have sought an elusive elixir from the blood of people with AIDS. There has been a desire – abetted enthusiastically by people with HIV, who would prefer to measure laboratory abnormalities than lesions, weight loss, colony counts and fevers – to find the perfect marker in the peripheral blood, the prognostic holy grail of pathogenesis. Many candidates were screened, many samples assayed. CD4 laboratory measurements were standardized, p24 antigen kits distributed to study sites, acid-dissociation methods promulgated, beta-2-microglobulin and neopterin fractions scrutinized, aberrant cytokines measured. Ironically, viral quantitation has proved, so far, less prognostic either of response to therapy or of outcome than immunological measurements such as CD4 count, B2M or elevated TNF and/or IFN-alpha. What these observations should have clarified is that disease progression and outcome depends on the status of the immune system, not just on the viral load. [According to Ho, viral load is highest during ARC.] Yes, the virus triggers something, and yes, this something results in the destruction of the CD4 lymphocytes, widespread immunologic havoc, and tissue destruction initially confined to lymphoid organs and later distributed throughout the body. But what is this black box, this occult destruction both of immune tissues and of immune cells, which occurs quietly somewhere for years and decades, then erupts into the blood and blossoms throughout the body with its gruesome efflorescence of clinical complications.
The preliminary consensus about the relevance of certain surrogates such as CD4 levels may have been beneficial from a regulatory point of view, since it enabled licensure of new treatments similar enough to AZT to justify the supposition that if they too raised CD4 counts they may confer a slight, sometimes measurable, later clinical benefit. But from a pathogenetic standpoint, this consensus was premature. Researchers were relying exclusively on the blood to develop their ideas. They were looking where the light was. This provided a false sense of (in)security about our understanding of AIDS. Every month, thousands of us anxiously scrutinize our CD4 counts, worrying over what may be artifacts of measurement or diurnal variation. The real damage is hidden away somewhere else. The real lesions of HIV infection are sequestered from the blood, and we do not even yet know whether what the blood tells us is an accurate reflection of what damage is occurring elsewhere.
The only way to find out what we must know is to adapt our approach to find out as much as possible as fast as possible. Basic experiments should be run on clinical samples alongside laboratory cell lines and virus isolates. No lab line or animal model will tell us with certainty whether a given mechanism is operative in people. Only clinical research – of a kind not yet being conducted – can tell us this. Only clinical basic research can help to unlock the pathogenesis in time to help us.
When I say basic researchers need to become more clinical, I mean that they must direct their efforts more to people with HIV and what is going on in our bodies, rather than exclusively in elegant and often artificial laboratory and animal models. In any case, SCID-hu mice don’t get sick. Neither do chimpanzees. Pigtail macaques get a primary viral syndrome, but we won’t know for years whether they develop AIDS. In any case, there aren’t enough pigtailed macaques to go around. There’s a shortage of pigtailed macaques. [I’m not saying that animal research isn’t vitally important, only that it isn’t enough.] Needless to say, there’s no shortage of people with HIV. We’re just still not yet being studied properly.
3. Activists and basic researchers need to work together.
Working together will not always be easy, but it need not be as difficult as was our initial engagement with the clinical research establishment. The differences here are as telling as the similarities. In both cases, we feel that AIDS research could be faster, more effective and more ethical if it involves us. In both cases, we feel that our perspective as people living with AIDS or HIV and those who cared for us provided an invaluable ethical and human impetus to the work.
Yet clearly basic research is different from clinical research. The cultural barriers may be higher. On the other hand, some basic researchers have told us that they feel neglected. Fear no more! Activist strategies which worked for clinical research will have to be radically altered if we are to affect basic research. Activists’ claim to expertise in clinical trials came out of lived experience. Most of us cannot claim the same for basic biomedical research. We can, however, hope to serve as catalysts for better and more coordinated work and as agitators for increased funds.
There are cultural obstacles as well as conceptual and practical ones. For starters, the “social contract” between basic researchers and people with HIV is less well defined. The parameters of the interaction are unclear. It is hard to get what you want if you’re not sure what you’re entitled to. Just as we had to struggle to obtain the most basic rights and dignities as clinical trials subjects, so if we offer to participate in basic research we shall have to develop strategies for protecting ourselves.
This spring, I carried out an N of one trial on myself to see how it felt being a subject of basic research. I probably became HIV positive sometime in the mid-1980s. I’ve monitored my CD4 count since mid 1988, when my ratio was 1:1. My CD4 levels have fluctuated in a virtual sine wave from 750 to as high as 1100 and to as low as 550 in the last four years. My most recent count was 660. While my CD4 levels surfed on the waves, my CD8 cells rose steadily from 850 (in mid 1988) to over 2000 today. Are these good anti-HIV cytotoxic T lymphocytes or bad autoimmune, anti-CD4 CTLs, or some combination of the above? Every lab value has at least two, and sometimes more, contradictory interpretations). I’ve also had intermittent lymphadenopathy. Yet at my CD4 level there’s certainly no compelling evidence to use AZT or any other nucleoside analogue, and I don’t live in a city where the trials of active immunotherapy with gpl60 are ongoing – even if I wanted to inject a risky immunogen into my system, with the possible attendant dangers of immune activation and the generation of cross-reactive anti-self or HIV enhancing antibodies. There were no trials for me. I wasn’t enthusiastic about the drugs available at the local drugstore. What could I do that might be useful?
I thought, last year we were demanding that they do tissue studies of HIV infection, why not put your body on the line? (After my sixth arrest last January at Astra failed to induce them to lower the price of Foscarnet from $25,000 a year, I had to think of some way to put my body on the line!)
In April I went up to St. Luke’s, subjected myself to local anaesthesia and a rather dreamy sedative which kept me babbling throughout the operation, and had one of my left axillary lymph nodes removed (that’s under the left armpit). The operation was brief and somewhat painless, and the wound healed quickly. They told me later the node was as big as a walnut. My tissue samples were sliced and preserved and placed on slides and sent to various laboratories up and down the East Coast for analysis.
I went through several weeks of preoccupation with what they would find. Of course, I wanted to know the answers to the pathogenesis questions. Of course, I wanted to know what was going on inside my body. It crept up on me in a somewhat devastating way. My doctor called me every few weeks to fill me in. “They’ve found a signal.” Oh. A signal of what? Later, “it’s crammed with virus.” Did they have to be so direct? Of course, something direct was what part of me wanted. But not “crammed with virus.”
Two weeks ago I finally got ahold of the results. Several slides with PCR analysis comparing the viral load in my blood versus my lymph node, and several pictures of my node stained with various techniques. The PCR was fairly routine. I’ve always objected to being reduced to a blob of nucleic acid on a film, so I did not bring a copy of my PCR to show you today. As you saw last year in Dr. Fauci’s presentation, the viral load in the peripheral blood – mine, as well as in the samples he showed last year – is one to two logs lower than in the lymph node. It’s hard to get too excited or upset about a blob of nucleic acid, however.
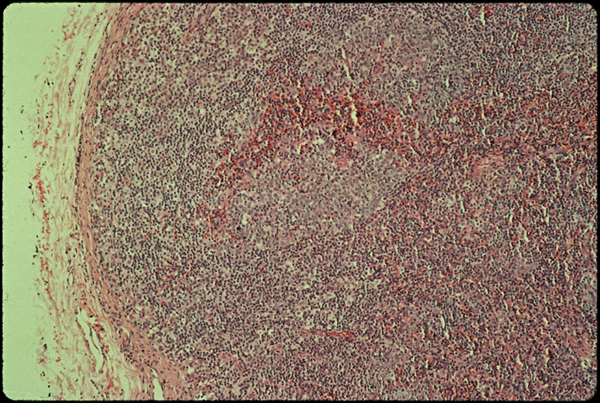
Slide one.
The tissue pictures were something else again. On light microscopy stained with H+E (slide one) there’s nothing too exceptional about me. The lymph node is filled with young lymphocytes, and the germinal centers where B cells mature are looking rather plump. They are active. Something’s turning them on.
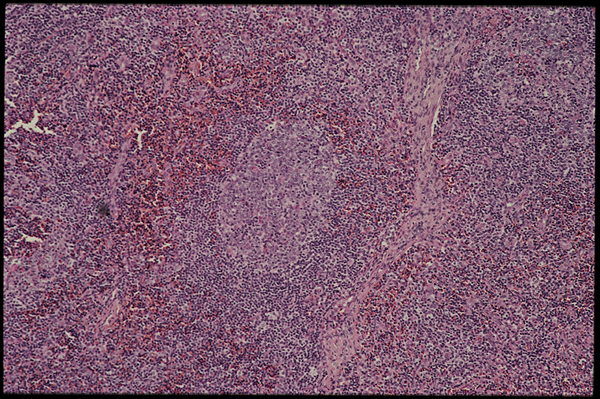
Slide two.
That’s a germinal center in the middle of this slide (slide two). These pictures were taken by Donald Kotler of St. Luke’s in New York.
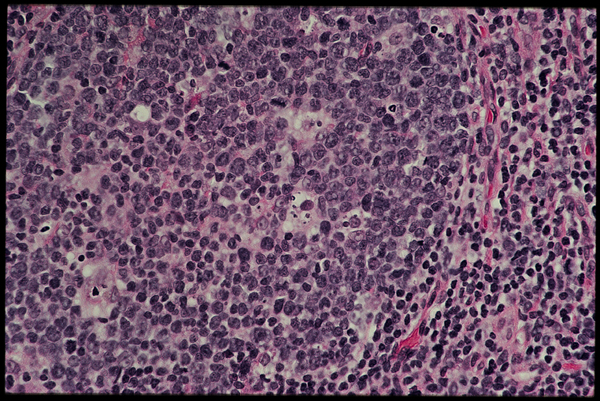
Slide three.
A closer look (slide three) reveals a lot of large, immature B lymphoblasts along with some smaller more mature B cells [and one or two possibly undergoing apoptosis?].
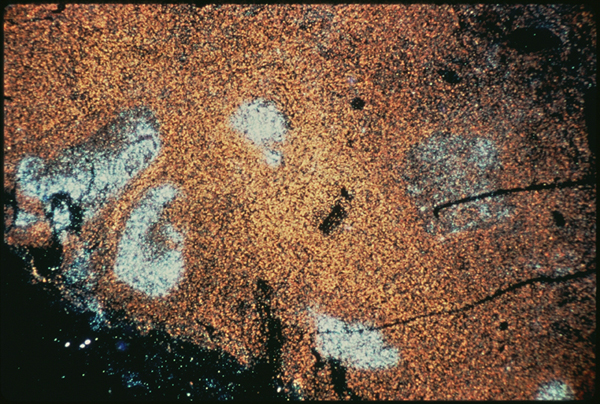
Slide four.
When my biopsy was stained with a DNA probe using in situ hybridization, however (slide four), the results were dramatic. This slide was taken by Dr. Cecil Fox of Yale University for Giuseppe Pantaleo and Anthony Fauci of the Laboratory of Immunoregulation at NIAID. If I understand correctly, the probe uses a complementary DNA strand which binds to HIV messenger RNA that is being produced inside of infected cells at the time the sample was fixed. Dr. Fox developed a method to protein digest the sample before hybridizing, and the viral signal is much clearer after protein digestion. This means that previously the probe could not detect viruses, presumably because the latter were obscured by antibodies. I believe that this particular probe recognizes HIV mRNA to gag coding sequences. That means this probe does not detect latently infected cells, it only labels cells which were actually making virus or virus proteins at the time I had my operation. In this case, the stain is bright over several germinal centers in the lymph node. It really was crammed with virus. It was very hard to look at this and think of me. This in me. All those old questions about “what causes AIDS if there are so few infected cells in the peripheral blood” sort of wilted away at that point. On the other hand, all those suspicions that the experts really know very little about what’s really going on in our bodies reared their ugly heads again. HIV on FDC in the B cell region? How does that fit into the T cell reservoir/T cell killer theory?

Slide five.
A second picture (slide five) is a close-up of a single germinal center. [The germinal centers are a matrix of follicular dendritic cells which display antigens or antigen-antibody complexes – in this case HIV – to immature B lymphocytes so that the latter may generate appropriate antibody responses.] What is remarkable is that most of the HIV is in the B cell area, not the T cell area, where you might expect it. Virus localized to the B cell area, and we know very little about how B cell hyperactivation contributes to T cell deletion and progression to AIDS. Note the central area of the germinal center. It is not stained as brightly. It may be undergoing infiltration by cytotoxic T lymphocytes or phagocytes. At some unknown stage of HIV disease, people’s lymph nodes go from swollen and active like mine to shrivelled and shredded. What destroys the follicular dendritic meshwork which supports the lymphoid tissue? Is it the body’s own reaction to the virus which stains so brightly there? Is the final CD4 decline only a “surrogate marker” for the final destruction of the dendritic reticulum which supports the staging and amplification of antigen-specific immune responses?
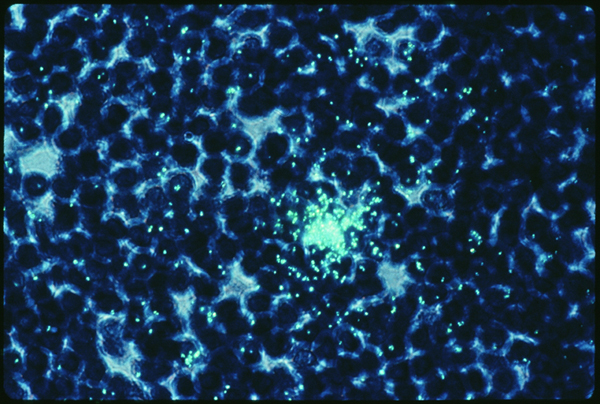
Slide six.
In contrast to the germinal centers B cell areas, only isolated CD4+ T cells are seen as infected (slide six). Here is a T cell in the paracortical region of my lymph node. I believe it is stained with the same in situ hybridization probe at a higher resolution and with a different dye. Clearly this T cell is making virus. (The aura of green marks on the outside of the cell are not RNA, they are the trace of the radioactive label as it hit the emulsion on the slide.) So there’s my T cell, infected with HIV. Who knows what would have happened to it if I hadn’t had my operation. Would it have churned out virus? Would it have burst? Would it have formed syncytia with other uninfected CD4 cells? Would it have developed anergy? Would it have become dysfunctional? Would it have attracted anti-HIV antibodies and been lysed by complement, or attracted anti-HIV CTL and been zapped? Would it have been fine until it met its programmed antigen, then undergone apoptosis?
I have all kinds of questions about what is happening to the cells and tissues of people with HIV and AIDS which are not being answered by basic research using lab lines and artificial HIV isolates. I have all kinds of questions about what is happening to the other types of immune cells and tissues in people with HIV which are not answered by assays which look exclusively for the virus. Cells and tissues and organs are affected by HIV and by the resulting immune deficiency, but all too often these effects are ascribed mindlessly to direct action of the virus. A progression of irrelevant papers document the in vitro infection of an enormous variety of cell types. What is happening in vivo? Cells are affected by these processes whether or not they are infected. What copathogens are involved in chronic immune activation? Does EBV play a role in stimulating B cells, and through IL-6, stimulating HIV? What immunoregulatory feedback circuits are disrupted? The list is endless.
So what did I get out of my little N of one experiment? Scared, confused and upset, partly. Graphic confirmation of something serious going on in my body, partly. A taste for how difficult it must be to be a doctor unable to recommend a therapy because none are indicated, and few under study. Certain parts of my body are crammed with virus. There is no treatment licensed for people at my stage of HIV infection. The tissue damage is localized. But no one could say I was in a latent stage of infection. My immune system appears to be holding HIV in check at least for now, but what about that infiltration in the germinal center? Will nucleosides lower virus production there? Nucleosides do not affect virus production in any chronically affected cell. They only prevent certain uninfected cells from being infected. Will active immunotherapy with viral peptides enhance my ability to clear these antibody antigen complexes? Active immunotherapy might only hasten the destruction of my germinal centers by stimulating new CTLs to attack those prominent viral targets. What cytokines could be detected in the lymphoid compartment? Are any cytokines being expressed which activate virus production? Is there IL-6 or TNF-alpha in my lymph node? (No one looked.] Is there Epstein-Barr virus (EBV) activity? [No one looked.] Does EBV induce B cell activation cytokines which participate in the induction of HIV and in the destruction of lymphoid tissue?
There are a lot of other questions which could be answered by further investigations of my tissue and that of other volunteers. But in future, I recommend you negotiate in advance what you are going to get from the encounter, because otherwise you’ll end up feeling peculiarly exposed and uncertain. I want to thank the investigators who made these pictures available to me. I know that this is a type of collaboration with which they (like me) are still unfamiliar. Thank you, Donald Kotler, Fred Kimmelstiel, Giuseppe Pantaleo, Cecilia Graziosi, Cecil Fox and Anthony Fauci. I hope it helps. This brings me to my final point.
4. Basic clinical research can simultaneously answer pathogenetic questions and speed therapeutic progress.
The ultimate treatment regimen for HIV-associated immune deficiency will probably have three components: OI treatment and prophylaxis, immune-based therapy, and antiretroviral therapy. We have made substantial progress towards the first (at least for treating the 5 most common OIs, and for preventing two), and some more debatable progress towards the third, with our limited armamentarium of short-term nucleosides. It’s my belief that antiretroviral treatment per se will remain inadequate until there are effective agents inhibiting virus production in chronically infected cells, since any RT inhibitor will eventually be overcome by resistant viral mutants.
But the second area I mentioned, that of immune-based therapy, remains an underfunded and ignored orphan of AIDS research. Yet I believe well-coordinated parallel investigations of basic and clinical immune-based interventions could help simultaneously to unravel the mysteries of the pathogenesis of AIDS and to speed the development of effective therapy.
The ACTG 160 study of pentoxifylline is one example of an intervention seeking to simultaneously determine the true incidence of TNF in PWAs and to see whether administering a TNF inhibitor reduces a) TNF and b) HIV load in PWAs. [CRIA in New York is now enrolling participants in a second, placebo controlled study of PTX which I wrote last year to confirm the results of ACTG 160.]
My basic point is that small, pathogenesis directed clinical studies carried out in close collaboration with HIV infected participants can help to shed light on the competing mechanisms far more rapidly than can traditional development from lab lines to mouse, monkey and ape, with extensive preclinical toxicology and pharmacology, drug scale-up, and finally, years later, pitifully slow, unimaginative and inadequate phase 1 clinical trials. Many of the potential interventions we have identified are already in common use and some are available by prescription.
Different mechanisms and different theories have different therapeutic implications. Possible modalities include Thl cytokines early (IFN-gamma, IL-2); plasmapheresis or other immune-complex clearance agents; immune deactivators such as CsA, FK-506 or anti-CD28 antibodies; IL-6 inhibitors; IL-10 inhibitors; CTL expansion (either polyclonal or HIV-specific); active immunotherapy with whole virus or HIV proteins or epitopes (presented with various vectors + adjuvants); passive immunotherapy; DHEA; anti-TcR antibodies; calcium channel blockers; other cytokine inhibitors (e.g., pentoxifylline, NAC); “classic” antiretroviral modalities; anti-infectives which combat opportunistic copathogens; and safe sex to reduce overall antigenic load.
Rapid progress can be made on both the pathogenetic and the therapeutic front by synthesizing the needs of activists and researchers. Basic research can become more clinical. Activists can help make this happen. In return, basic researchers must figure out how to work with people with HIV. Thank you.