JULY 2012
By Polly Clayden
NOTE: a postscript section at the end of this article includes periodic updates since publication in June 2012.
In June 2010, the WHO and UNAIDS launched Treatment 2.0, a strategic approach to the achievement of universal access to antiretroviral therapy (ART) and to making the most of the role of ART in preventing new infections.[1] A critical component of the strategy is the development of optimized, simpler, less toxic, and more efficient antiretroviral (ARV) drug regimens. It includes establishing optimal doses of ARVs (including possible dose reductions of existing ones), reducing pill count, using fixed-dose combinations (FDCs), and expanding access to safe, effective, and affordable drug regimens.
The ideal characteristics of a “dream regimen” have been variously described, and the target is one that is “so safe, effective, tolerable and durable that the need for switching to a new regimen would be very rare.”[2],[3],[4]
TABLE 1. Target Product Profile of a Dream ARV Regimen

A dream regimen, which encompasses all the characteristics of the target product profile, is a few years away but we might be able to do better with what we have already. We should also keep a close watch on what’s on the horizon. This new chapter explores the ongoing research into treatment optimization including dose optimization with existing compounds, and possible future opportunities with new ones at the nearer end of the pipeline.
Discussions about dose optimization—particularly through appropriate dose reduction—of approved antiretrovirals have been ongoing now for over a decade,[5],[6] the rationale being that when developing new drugs, the highest tolerated doses in phase II are often selected for phase III and, in turn, approval, where in some cases lower doses may have equivalent efficacy. Opportunities with some of the currently approved ARVs could offer several advantages over the approved doses:
- Reduction of the active pharmaceutical ingredients (API) used in a compound could lead to reduction in price (API accounts for approximately 70% of the price of generic ARVs);
- Potential reduction in toxicities; and
- Reduction in volume could make co-formulation easier (in resource-limited settings, 80% of people are treated with FDCs).
Research into these strategies has gained momentum recently, including endorsement by the WHO/UNAIDS as part of Treatment 2.0. In addition, the Clinton Health Access Initiative (CHAI) has undertaken the execution and coordination of a number of projects, and the Bill & Melinda Gates Foundation is providing substantial donor support.[7],[8] Several dose optimizations of antiretrovirals, including a number of clinical trials looking at dose reduction, are under way or in discussion.
Dose optimization strategies
There are several ways in which dose optimization might be accomplished:
- Dose reduction. In order to achieve regulatory approval for a dose lower than that currently approved, fully powered non-inferiority studies (phase III)—similar to those conducted by industry for the approval of a new drug—need to be done. It would take about three to six years to generate sufficient data to file with regulatory agencies, plus time to approval (about three months to a year). The estimated cost would be US$15–22 million.
- Reformulation. This strategy makes use of technologies and/or inactive ingredients to increase the bioavailability of a drug. A reformulated compound will need bioequivalence studies with the approved formulation (phase I). The estimated time frame to regulatory filing is two to three years, at a cost of US$2–8 million.
- Process chemistry. It may also be possible to alter the manufacturing processleading to more efficient and less expensive API production. For this strategy to be successful, regulatory authorities would need to see only equivalent stability and purity data. This would take about one to two years, at an estimated cost of US$1–2 million.
Source: Crawford KW, Brown Ripin DH Levin AD, et al. Optimising the manufacturing, formulation, and dosage of antiretroviral drugs for more cost-efficient delivery in resource-limited settings: a consensus statement. Lancet Infect Dis. 2012;12(7):550–60.
Tenofovir
Tenofovir is preferred as part of first-line treatment (in combination with lamivudine and efavirenz). It is broadly considered to be the best NRTI (nucleoside reverse transcriptase inhibitor)/NtRTI (nucleotide reverse transcriptase inhibitor) on the market, and this is likely to continue for several years.
In recent years, its price has dropped considerably. A tenofovir-based FDC regimen is now available at an annual per-patient cost of less than US$159.* There are, however, limits to tenofovir’s lowest possible price due to its high milligram dose (300 mg) with the current formulation. This also makes it less easy to co-formulate with other antiretrovirals.
CHAI is currently working on reformulation of tenofovir in partnership with a generic manufacturer. Although the new dose has yet to be determined, the researchers anticipate a reduction by about a third.
Additionally there are two new pro-drugs of tenofovir in development: GS-7340 (steaming ahead) and CMX-157 (almost moribund), which Simon Collins describes elsewhere in this report.
* All prices quoted in this chapter are from the CHAI ARV Ceiling Price List, andthe Médecins Sans Frontières (MSF)Drug Prices & Patent Status list.
Zidovudine
If tenofovir remains the preferred first-line NRTI/NtRTI, zidovudine is likely to be used second-line in the short term.
Although zidovudine is generally better tolerated than stavudine over a long-term period, its hematologic toxicities (anemia/neutropenia) remain a concern in many resource-limited settings (RLS).
The ongoing MINIZID study is looking at 200 mg versus 300 mg zidovudine twice daily (as part of a regimen with lamivudine plus a non-nucleoside reverse transcriptase inhibitor [NNRTI]), with reduction of anemia as the primary endpoint. This is a 48-week phase II study in 136 treatment-naive patients, sponsored by the University of Geneva and being conducted at the Hôpital de la Caisse Nationale de Prévoyance Sociale, Yaoundé, Cameroon. Recruitment began in August 2011.[9]
The study will not generate sufficient data for regulatory approval of the lower dose, but will provide proof of principle.
Some Asian countries such as Thailand and India already use the zidovudine 250 mg tablet twice daily, and Thailand is already using 200 mg twice daily in patients weighing less than 50 kg.
Stavudine
Of all the dose optimization strategies proposed or ongoing, the decision to use stavudine is the most controversial. Unlike the other antiretrovirals for which these strategies are being suggested or conducted, stavudine is no longer a preferred option due to its toxicity profile.
A proposed phase IIIb study plans to compare 20 mg stavudine twice daily to 300 mg tenofovir once daily in approximately 1,000 patients. The primary objective is to demonstrate the non-inferiority of stavudine to tenofovir (both in a regimen with lamivudine plus efavirenz) in treatment-naivepatients. The proportion of patients in each regimen with undetectable viral load (<200 copies/mL) at 48 weeks would determine this.
The secondary endpoints are to evaluate the tolerability, overall safety, and efficacy of 20 mg stavudine compared to tenofovir.
The trial would be conducted at sites in India, South Africa, and Uganda and sponsored by the Bill & Melinda Gates Foundation.
This trial is concerning, as it will not answer stavudine’s long-term toxicity question. The 20 mg stavudine dose might be acceptable in a short-term 48- or even 96-week virological endpoint study. However, because mitochondrial toxicity is both dose- and time dependent, many of stavudine’s most serious side effects (such as peripheral neuropathy and lipoatrophy) would not necessarily emerge until after such a study was completed. Although it looks at lipoatrophy, this study does not include monitoring of surrogate markers for mitochondrial toxicity, so it cannot shed light on the incidence of this serious adverse event.
The stavudine parallel track program, which randomized over 10,000 patients to receive 40 (30) mg or 20 (15) mg (between October 1992 and February 1994), showed a higher incidence of neuropathy in the high-dose arm (21%). Nonetheless, the incidence of neuropathy observed in the lower dose arm was also unacceptably high (15%).[10]
In addition to concerns about cumulative toxicities, stavudine-related cost savings might become irrelevant by the trial’s end. Through other dose optimization strategies and the expected approval of promising pipeline compounds (such as GS-7340 and douletegravir), alternatives are likely to become available in a similar time frame that could drive regimen costs down with less risk to patient safety.
Importantly, stavudine is extremely unpopular with people with HIV and activists.[11],[12] For example the Malawi Network of People Living with HIV/AIDS (MANET+) held a press briefing concerned by the slow pace for phasing out current use of this drug in Malawi. Despite the funding crisis the Malawi government has prioritized this to be completed by June 2012.[13]
In South Africa and India, people with HIV and activists are preparing protests and petitions against the trial and the slow phase out of stavudine.[14],[15]
As Bad Science’s Ben Goldacre asked: “Why is the Gates Foundation supporting this trial of arubbish AIDS drug?”[16]
Efavirenz
Efavirenz is currently the preferred anchor drug. Price and possibly central nervous system (CNS) toxicities could be reduced if a lower dose than the currently recommended 600 mg is possible.
The ENCORE1 study, which began recruitment in September 2011, is looking at 600 mg versus 400 mg of efavirenz in 630 treatment-naive patients. The ENCORE studies are designed to compare lower doses with approved doses of antiretrovirals. Pharmacokinetic studies of lamivudine and lopinavir (ENCORE2 and ENCORE3) have already been conducted as part of this program, with the conclusion that neither is a suitable candidate for dose reduction.[17],[18],[19]
The primary endpoint for ENCORE1 is the comparison between treatment groups of proportions of patients with viral load <200 copies/mL 48 weeks after randomization. The complete follow up is 96 weeks, and there are sites in Europe, Australasia, Latin America, Asia, and Africa. The trial is fully recruited, and results are expected in 2013.
ENCORE 1 has two substudies designed to look at pharmacokinetics (PK) and CNS exposure.[20],[21]
If successful, this trial will generate sufficient data to gain regulatory approval and change World Health Organization (WHO) and other key treatment guidelines.
There are concerns about the drug/drug interaction with rifampicin in TB/HIV coinfection if the efavirenz dose is reduced.
The high API of efavirenz is due in part to its poor water solubility. CHAI is in discussion about reformulation work to improve this.
Atazanavir
Dose reduction may also be possible with atazanavir, and the HIV Netherlands Australia Thailand Research Collaboration, with some support from the Kirby Institute, is conducting a trial that will provide some evidence for this strategy.[22]
The low-dose atazanavir/ritonavir versus standard-dose atazanavir/ritonavir (LASA) study is comparing the efficacy and safety of atazanavir/ritonavir at either 200/100 mg or 300/100 mg once daily in Thai patients in combination with two NRTIs. This non-inferiority, phase IV study with about 600 patients began recruiting in March 2011 and has a time line similar to that of ENCORE1.
This study is enrolling patients who are already virologically suppressed to switch to the lower or standard dose of atazanavir. This research is important for Thailand as patients tend to have a lower body weight, and hyperbilirubinemia occurs quite frequently. It will be difficult to generalize the results from this research beyond the study population, but positive results would provide good reason to conduct a study in treatment-naive patients from a broader population.
Atazanavir is also poorly water-soluble, and CHAI is looking at the possibility of reformulation.
Darunavir
Darunavir is generally considered to be the most durable protease inhibitor (PI), but there is no generic formulation, and cost has been a barrier to its wide use.
This drug also has different approved doses for treatment-naive (including treatment-experienced but with no darunavir-associated mutations) and PI-experienced patients. Treatment-naive patients receive darunavir/ritonavir at an 8:1 (800/100 mg) ratio once daily, and experienced patients at a 6:1 ratio (600/100 mg) twice daily. There may be potential for dose reduction to 400/50 mg.
The ratios also vary for children depending on their weight band and treatment experience.
The establishment of single ratios for adults and children would make simpler darunavir-based regimens and formulations more feasible.
CHAI is also looking at optimizing the formulation.
Ritonavir
It may also be possible to give atazanavir and darunavir with a lower boosting dose of ritonavir. Lower doses could be better tolerated, cheaper, and easier to co-formulate with PIs than the current dose.
If a 50 mg heat-stable tablet of ritonavir could be manufactured or 50 mg coformulated with either protease inhibitor, new bioequivalence trials would be needed to ensure that boosting effects were similar to those that have been achieved previously in small pharmacokinetic trials with the liquid formulation.
A 50 mg ritonavir tablet would also be very useful for pediatric dosing, as the liquid is expensive, impractical (particularly for resource-limited settings) and tastes dreadful.[23]
Lopinavir
The current tablet formulation of LPV/r is 118% bioavailable, compared to the original gel capsule formulation. Taking a regulatory approach using existing data could possibly be sufficient for the approval of a lower dose with this compound, and this strategy has been discussed. Although lopinavir is now the most widely used protease inhibitor, both atazanavir and darunavir are considered to be better options, so this approach may not be pursued as it is of low priority.
TABLE 2. Approved ARV Compounds with Potential for Dose Optimization
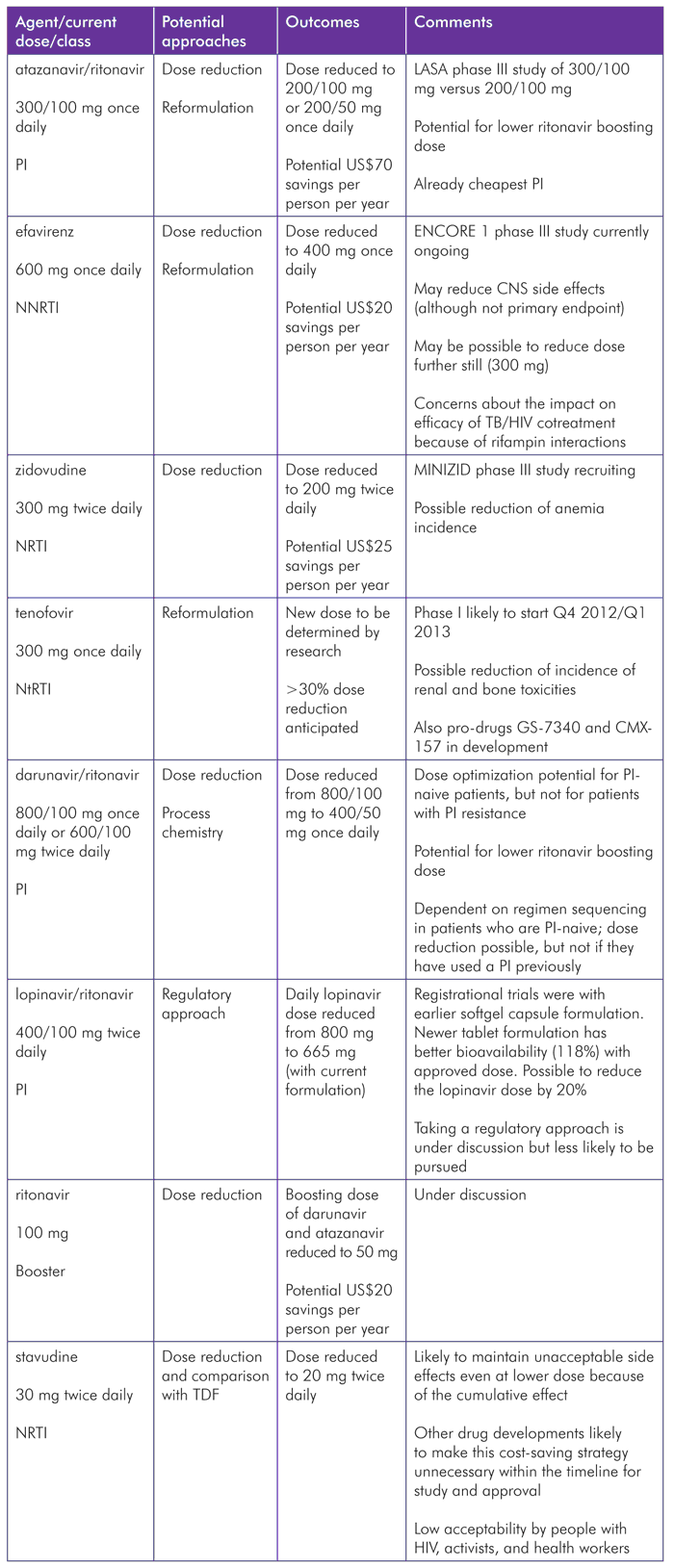
What to Expect in the Next WHO Guideline Revisions
It is expected that in the short term the preferred adult first-line treatment will remain an FDC of efavirenz/tenofovir/lamivudine, and that the next WHO guideline revisions will not be dramatic—and reports from recent expert meetings reflect this.[2],[3],[4],[24],[25]
Central nervous system toxicities that are a concern with efavirenz might be reduced with the lower dose under investigation in ENCORE1, and the trial includes a substudy to look at this aspect. Fears about its use during pregnancy are steadily being assuaged, and more permissive recommendations—in keeping with the most recent British HIV Association guidelines, and suggested in the recent WHO programmatic update on ARVs for pregnant women—are expected in the 2013 guidelines.[26],[27],[28],[29],[30]
Despite direct comparisons, lamivudine and emtricitabine are largely considered to be interchangeable in terms of efficacy and safety, and a recent WHO systematic review concluded this to be true.[31] Both are NRTIs and are structurally similar molecules with low toxicity, and both are effective against hepatitis B virus. Cost comparisons make lamivudine the preferred option—using emtricitabine instead in combination with efavirenz and tenofovir adds US$24 per patient per year to the cost of a first line regimen FDC, and US$27 to a combined product with tenofovir.
Work on the bioavailability of tenofovir could bring down the price (currently US$58 as a single agent), and further reductions still might be possible with the new pro-drug.
Boosted protease inhibitors plus two NRTIS are recommended for second-line treatment, and this is not expected to change in the short term. The FDA has recently tentatively approved a heat-stable formulation of atazanavir/ritonavir.[32] This 300/100 mg one-pill once-daily formulation is US$276 per patient per year and compares favourably to heat-stable lopinavir/ritonavir costing US$378, with four pills a day and twice-daily dosing. Once-daily heat-stable boosted darunavir might also be an option, but at present a suitable formulation (and suitable price) remains elusive. Dose reductions of atazanavir, darunavir, the ritonavir booster, and zidovudine (which will be used second-line if tenofovir is used first-line) are all being investigated or considered.
Recommendations for third-line treatment were introduced for the first time only in 2010, and suggest boosted darunavir, raltegravir (the only approved integrase inhibitor), and etravirine (second-generation NNRTI) in nucleoside sparing regimens. Again, little change is expected beyond the possible expansion of options in the integrase class (boosted elvitegravir and dolutegravir). None of these yet have generic versions, and the cost is considerable. More detailed guidance is needed in the next revision.
Opportunities with Pipeline Drugs—Ones to Watch
The integrase inhibitor dolutegravir, currently in phase III, with expected approval in 2013, is a compound with high potential, and it is predicted to cost US$30 per patient per year: 90% cheaper than raltegravir.**,[33] It is a small molecule (50 mg), compared to elvitegravir (150 mg once daily plus 150 mg cobicistat) and raltegravir (400 mg twice daily), with once-daily dosing in treatment-naive patients. Early data suggest that a dose increase (to 50 mg twice daily) will be needed with TB treatment.[34] Dolutegravir appears well tolerated, and with the potential to be low-cost could potentially replace efavirenz first-line or be used second-line. Trials in children, including in neonates, are planned and a granule formulation is in development.
** More extensive details and references for the investigational antiretrovirals are provided in the ARV chapter of this report, and for their respective investigational plans in children in the pediatric ARV chapter.
Further down the pipeline, but also with high potential, is the tenofovir pro-drug GS-7340. An interaction with cobicistat makes it possible to use a 10 mg dose when it is co-formulated with this boosting agent.[35] The dose is still to be announced for the single agent, but is expected to be 25 mg.[36] With doses 10 times or more lower than that of the existing formulation of tenofovir, the cost of GS-7340 is predicted to be appropriately lower, and could come in at an annual patient cost of as little as US$20.*** A question with this compound is whether increased intracellular concentrations of GS-7340 accumulate in renal tubule cells and, in turn, cause associated toxicity. No renal problems were observed after 10-day exposure, but this is an important aspect of further studies. It is unfortunate that this compound was not prioritized for development earlier, as in vitro data were presented 10 years ago.[37]
*** i-Base/TAG estimate based on fixed cost of TDF API, inactive ingredients, and packaging.
With the potential to completely alter standard of care, discussions about, and early development of, long-acting formulations are also under way for monthly or weekly depot injections. Potential candidates might be rilpivirine and GSK1265744, both in early stages of development, plus CMX-157, which also has a long half-life, but the future of this molecule is currently unclear.[38],[39],[40] As yet, though, we do not have clarity on the target product profile, nor is it clear if the right combination of drugs required to construct a suitable regimen are available or even in development.[3]
Discussion
The ARV chapter of this report describes a number of FDCs, either filed with the FDA or in phase III, targeted at markets in industrialized countries. These are either “incestuous” combinations of compounds from the same manufacturer, e.g., Quad, Quad 2, and 572-Trii, or licensing agreements between companies where there is no competing alternative component, such as that between Gilead and Janssen to formulate darunavir/cobisistat/emtricitabine/GS-7340. And, as noted in that chapter, “virological failure with resistance to one FDC is likely to preclude reliance on others.”
Gilead, Janssen, and BMS are also investigating cobicistat with darunavir and atazanavir as co-formulated boosted PIs, although it is unclear whether cobicistat offers any advantages over ritonavir.
With respect to RLS, Quad is not expected to become a preferred option, with dolutegravir on the horizon, elvitegravir requiring a boosting agent, and lamivudine preferred to emtricitabine. 572-Trii is also not entirely appropriate as the cost of abacavir and concerns about hypersensitivity have meant this NRTI is not recommended or widely used (except in pediatric treatment). The darunavir-based FDC is targeted at first-line patients (with 800/100 mg darunavir/ritonavir), and so is also unlikely to be a useful option according to current (and expected short-to-medium-term) guidance, sequencing discussions, and pricing.
Although intellectual property is not the primary focus of this report, Table 3 shows pipeline FDC products and their respective patent information, and illustrates the hurdles that would need to be overcome were these FDCs, or others, to be produced by generic companies for RLS. The Medicines Patent Pool, which negotiates with the innovator pharmaceutical companies to license their drugs through the pool so that generic companies can then access these licenses, and in turn produce cheaper versions, seems the most promising mechanism to make newer drugs affordable and produce FDCs when patents are held by different companies.[41]
TABLE 3. Pipeline Combined Products including FDCs, and Patent Information
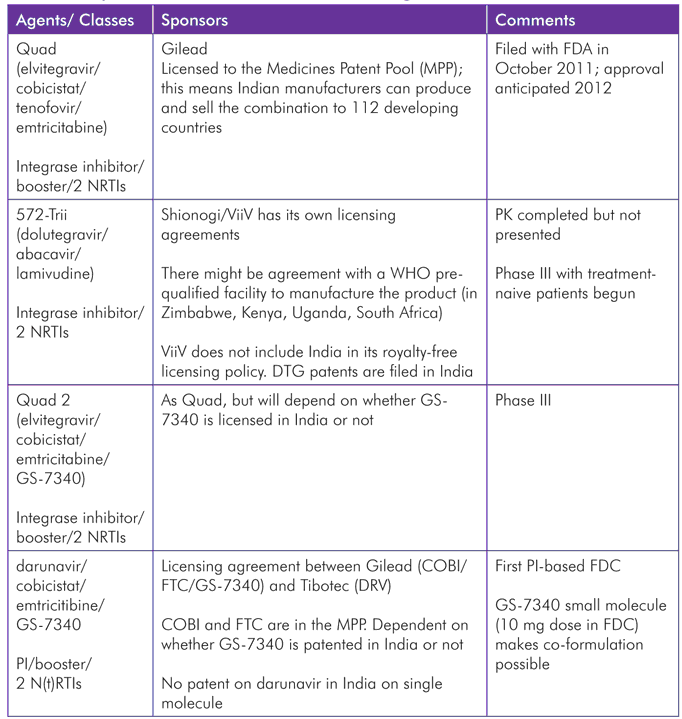
Source: Updated from the 2011 MSF antiretroviral sequencing meeting report: http://www.msfaccess.org/sites/default/files/MSF_assets/HIV_AIDS/Docs/AIDS_Event_SequencingMtg_Report_ENG_2011_FINAL.pdf.
What Needs to Be Done?
Treatment optimization must be in the interests of people with HIV. Seeking a comeback for a drug virtually abandoned in rich countries, for the sake of cost, and against much opposition from people with HIV and activists, is unacceptable. It is unclear why the Bill & Melinda Gates Foundation—which plans to fund the trial to look at stavudine 20 mg—consider this study to be a priority as it also does not fit with the one-pill, once-a-day target regimen. As we have written elsewhere, it seems an aberration in an otherwise carefully considered strategy for supporting research into the optimization of ART for RLS. This includes the ENCORE1 study of low-dose efavirenz, the reformulation of tenofovir to increase its bioavailability (working with CHAI), and the development of innovative, potentially long-acting formulations.
Drugs and regimens need to be designed with RLS in mind. The target product profilehas been widely described by now. Currently approved and pipeline compounds fit for this purpose need to be produced in appropriate formulations.
Shorten time between full FDA/EMA approval and WHO prequalification, FDA tentative approval, and approval by local regulatory agencies. This is critical. As Nathan Geffen describes in his commentary, national agencies such as the Medicines Control Council (MCC) in South Africa often take many years to register new medicines.
Delays with the registration process, in addition to production by generic manufacturers and recommendations in national guidelines, means that it takes years from promising results in trials and initial approval to wide availability for the majority of people in need of antiretroviral treatment. Despite almost 150 single agents and combination products having FDA tentative approval, the majority are older drugs and those with expired patents.
Postscript updates
August 2012
In July, Chimerix signed a worldwide license with Merck for CMX 157, their novel pro-drug of tenofovir that has lain dormant for sometime (described as “moribund” here). [42] Merck, self described as “A global healthcare leader working to help the world be well” have yet to impress us with their access prices.
Price was the big story as soon as Quad (now Stribild) was approved by the FDA. [43]